New Kubeflow Version 1.9 deployed on MQS Infrastructure
MQS Documentation
This blog is now defunct. Head over to https://docs.mqs.dk
![]()
New Kubeflow Version 1.9
Kubeflow recently released Version 1.9 with a variety of feature improvements such as a centralized model registry, security enhancements, and integration/installation improvements. We have now deployed Kubeflow 1.9 in the MQS infrastructure and you can access Kubeflow with the Quantum & Machine Learning Tier: https://dashboard.mqs.dk/subscriptions
Here an overview of the important menu items under Pipelines which we currently utilize at MQS to design well thought out calculation pipelines:
- Pipelines
- Experiments
- Runs
- Recurring Runs
- Artifacts
- Executions
This allows us also to design the Cebule Application Programming Interface in a consistent way and to bridge MLOps together with quantum chemistry, quantum computing and machine learning tools.
Some of the changes between version 1.8 and 1.9 we found important with respect to navigating and applying our model library with Kubeflow:
The Central Kubeflow Dashboard UI has been improved, with all Kubeflow Pipelines links now being under the Pipelines tab as shown on the left below:
The UI has now been updated to support visualizing output artifacts such as the training loss plot from a MQS ML model fine-tuning pipeline:
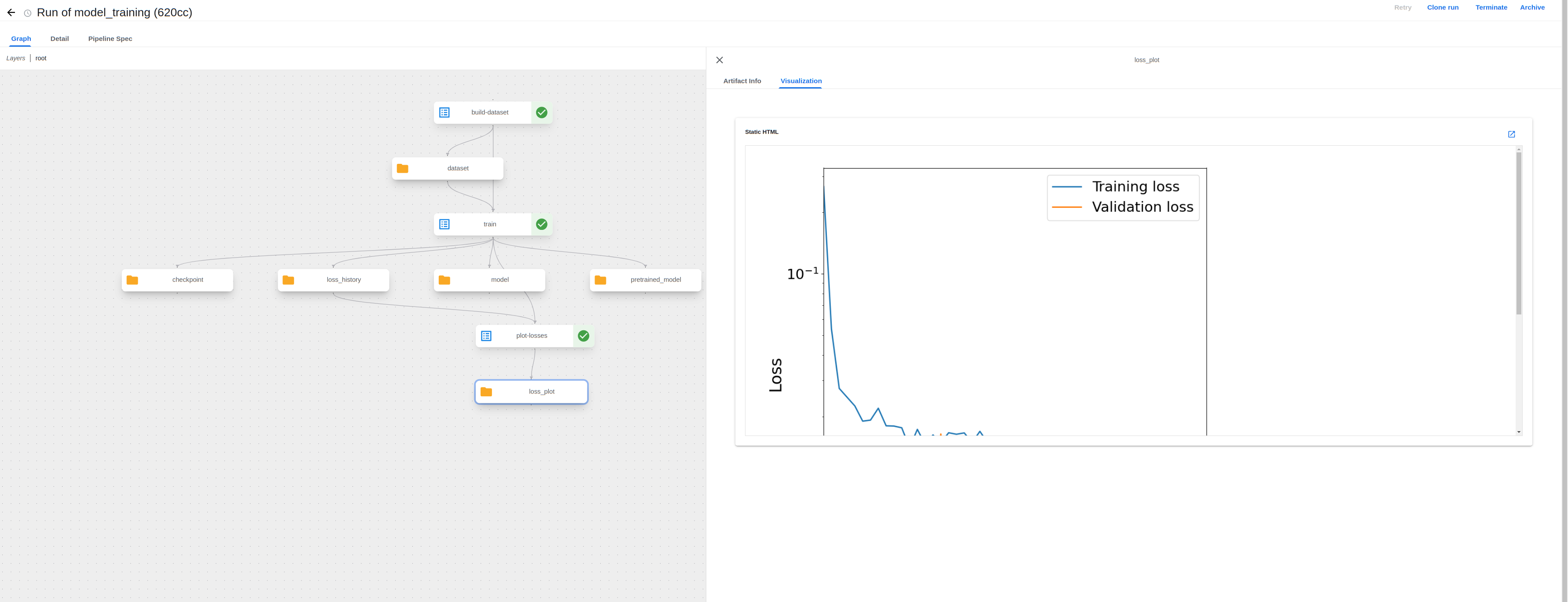
Visualizing images in the UI allows one to conveniently view output results (ex. evaluate a training run with the loss plot) without having to download the output files and view them locally, as was previously needed.
Applying the MQS Model Library
As part of our Quantum & Machine Learning Tier, we provide quantum chemistry and ML models for on-demand use such as geometry optimization with different methods, and graph neural network training/fine-tuning to predict molecular target properties.
These models can be accessed through Pipelines -> Shared on the Central Dashboard:
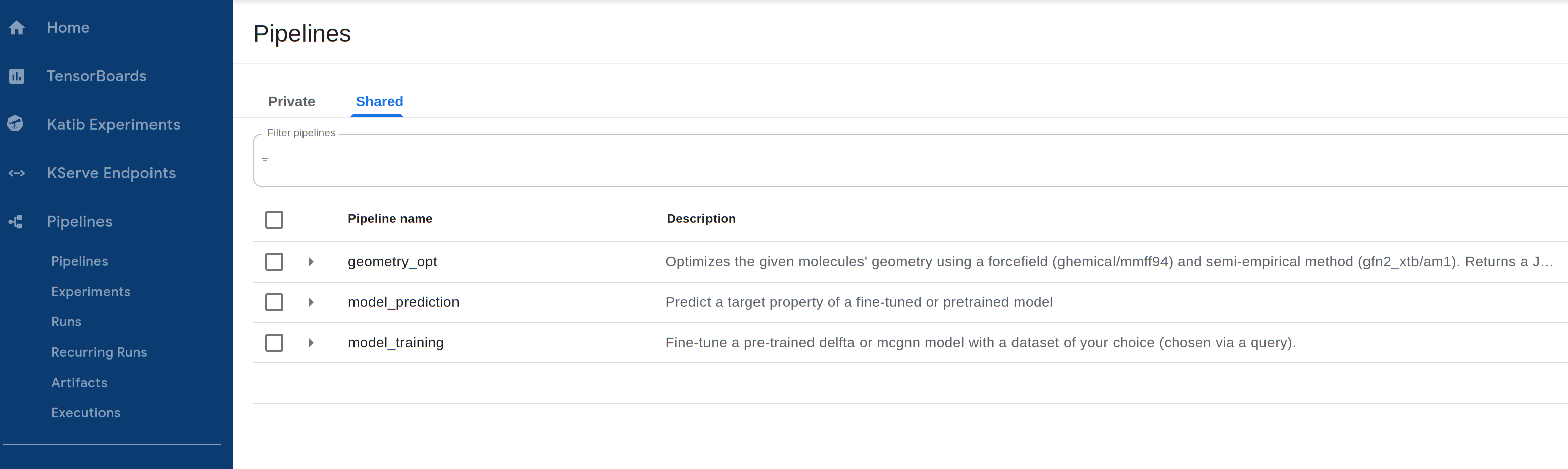
Additionally, the models included in the Quantum & Machine Learning Tier, plus the Enterprise Matrix-Completed Graph Neural Network (MCGNN) for generating Hamiltonians of molecules, are available through the pay-per-usage Cebule API and can be used in a Jupyter Notebook with our SDK as described in a previous tutorial.
The following models/pipelines are available, with more being continuously added to the model library.
Overview of Kubeflow Pipelines
geometry_opt
Semi-empirical geometry optimization of molecule 3D coords after initial force field optimization.
Inputs:
force_field: str from [mmff94, ghemical].
optimization_method: str from [gfn2_xtb, am1].
smiles_list: List[str] of SMILES as JSON str.
Cebule TaskTypes applied:
GEOMETRY_OPT
Artifacts:
None
Output:
List[List[Tuple[float, float, float]]] as JSON str containing each molecule’s optimized 3D coords.
model_training
Fine-tune a graph neural network on a custom dataset to predict a molecule property of interest.
Inputs:
model_name: str unique to this model (only letters, numbers, underscores).
model_type: str from [mcgnn, delfta].
query: str to select dataset molecules from MQS Database.
target_property: str for the model to learn to predict; currently [homo_lumo_gap] supported.
Cebule TaskTypes applied:
GNN_DATASET_CREATE
GEOMETRY_OPT
GNN_DATASET_EXTEND
GNN_TRAIN
Artifacts:
test_loss_mae: Metrics
loss_history: Metrics
loss_plot: HTML`
model: Model
pretrained_model: Model
Output:
None
model_prediction
Predict a property of interest with a pre-trained or fine-tuned model.
Inputs:
model_name: str to use for prediction; fine-tuned model or from [mcgnn, delfta] for pre-trained model.
smiles_list: List[str] of SMILES as JSON str.
Cebule TaskTypes applied:
GNN_PREDICT
Artifacts:
model: Model
Output:
List[float] in JSON containing predicted target property for given molecules.
Overview of Cebule TaskTypes (Python SDK)
GEOMETRY_OPT
Semi-empirical geometry optimization of molecule 3D coords after initial force field optimization.
Inputs
force_field: str from [mmff94, ghemical].
optimization_method: str from [gfn2_xtb, am1].
smiles_list: list[str] of SMILES.
max_processors: Limits concurrency of optimization.
Output
list containing each molecule’s optimized 3D coords.
GNN_DATASET_CREATE
Create a dataset for training or prediction.
Inputs:
dataset_name: str unique to this dataset (only letters, numbers, underscores).
target_property: str the dataset is meant for from [homo_lumo_gap, eigenvalue] where eigenvalue is for predicted hamiltonians.
includes_target_val: bool whether the dataset includes the ground truth target values.
max_processors: None
Output
None
GNN_DATASET_EXTEND
Add datapoints to an existing dataset.
Inputs:
dataset_name: str to add data points to.
molecule_chunk: Dict[str, Union[List[str], List[List[Tuple[float, float, float]]], List[float]]] containing the keys smiles, coords (for homo_lumo_gap datasets), and target_val, each containing a list of that data for each molecule.
max_processors: None
Output:
None
GNN_DATASET_GET
View a chunk of an existing dataset.
Inputs:
dataset_name: str to view.
start: int index to begin (inclusive).
end: int final index (exclusive).
max_processors: None
Output:
Dict[str, Union[List[str], List[List[Tuple[float, float, float]]], List[float]]] containing the selected datapoints.
GNN_DATASET_DELETE
Delete an existing dataset.
Inputs:
dataset_name: str to delete.
max_processors: None
Output:
None
GNN_TRAIN
Train a GNN to predict molecule effective Hamiltonians, or fine-tune a GNN to predict a molecule property.
Inputs:
dataset_name: str to train on.
model_name: str unique to this model (only letters, numbers, underscores).
model_type: str optional from [mcgnn, delfta] (default mcgnn, note that only mcgnn supports eigenvalue datasets).
hamiltonian_len: int optional: only for eigenvalue datasets, defaults to 128 for 128x128 hamiltonians.
hyperparameters: Dict[str, Union[float, int]].
optional keys:
epochs (default 100)
batch_size (default 8)
initial_learning_rate (default 2.0 * 10-3)
max_processors: Used to limit concurrency of data loading
Output:
Dict[str, Union[float, Dict[str, List[float]]]] containing the key test_mae for the MAE test loss, and loss_history with the training and val loss (MAE for eigenvalue, MSE for other properties).
GNN_PREDICT
Predict a target property or hamiltonian of molecules with a trained model.
Inputs:
dataset_name: str to run prediction on.
model_name: str to use for prediction (can be from [mcgnn, delfta] for pre-trained model, for any property other than eigenvalue).
return_upper_triangle: bool, optional only for eigenvalue datasets to return the flattened upper triangle of predicted hamiltonians instead of the full square matrix (default False).
max_processors: Used to limit concurrency of data loading.
Output:
Dict[str, Union[float, List[float], List[List[float]], List[List[List[float]]]]]: containing the key predictions for the predicted target values/hamiltonians/flattened upper triangle of hamiltonians,
optionally mae for the MAE of the predictions if the dataset includes ground truth target values.
Python SDK notebook examples
For a detailed example of running our Kubeflow pipelines, see our tutorial on the pipelines for training, fine-tuning and using the MCGNN and DelFTa ML models to predict molecule HOMO-LUMO gaps.
Further, we recommend to take a look at all Python-SDK example notebooks here: https://gitlab.com/mqsdk/python-sdk/-/tree/main/notebooks
We hope you find our integration of Kubeflow’s latest version and our Quantum Chemistry/ML models library can be of value for interesting studies with molecular quantum information.
You are always welcome to reach out if you have any feedback, questions, ideas or even collaboration projects to push this kind of work and research further (contact (at) mqs [dot] dk).