MQS Search API - Part 2: QMugs and PubchemQC PM6
MQS Documentation
This blog is now defunct. Head over to https://docs.mqs.dk
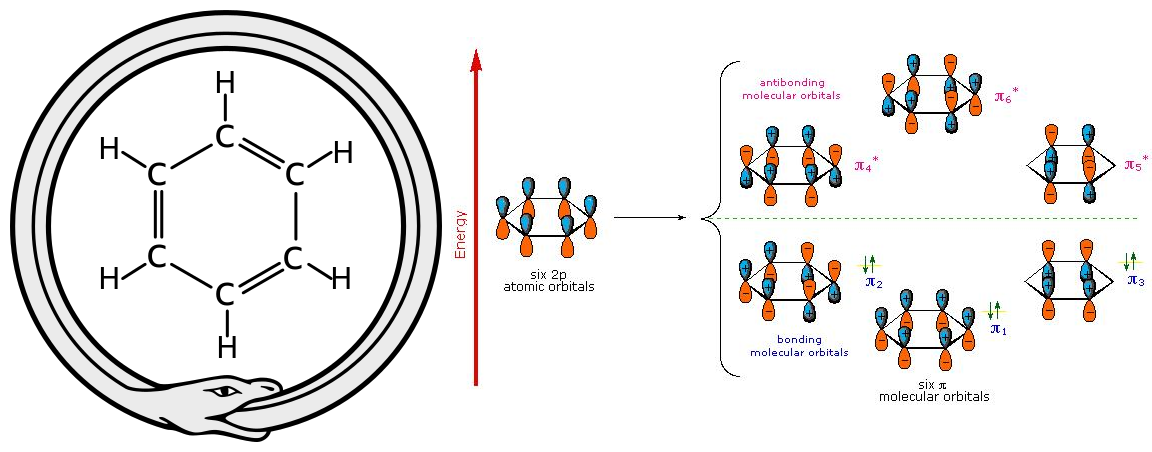
Figure 1 (left): An ouroboros with a Benzene ring. The famous dream or creative process by August Kekulé discovering the alternating double bounds in the Benzene ring structure (Source: Wikimedia).
{kind=link}
Figure 2 (right): The quantum chemical perspective: Cyclic overlapping of six p_z orbitals of the sp2 hybridized C-atoms forming a delocalized pi system (Source: LibreTexts Chemistry).
MQS Search API
In the MQS Search API Part 1 Tutorial we gave a basic introduction to the MQS Search API and will recap some details for getting an overview of the API.
Additionally, this Part 2 Tutorial will showcase how the Search field of the MQS Dashboard can be utilized to filter for molecules from the MQS database to then implement the defined search query via the Search API.
The database now also holds the QMugs data set with the community (UI for free) and basic subscription plans (API access + JupyterLab). In the end of this tutorial an overview of the PubchemQC PM6 and QMugs data sets is provided and we show how to retrieve data from the QMugs data set.
Overview of API endpoints
When interacting with a REST-API we can define sending data via the API with a POST call and receiving data with a GET call to the server. The following POST and GET methods exist currently with the MQS Search API:
POST /token: This endpoint generates an access token that is required to call the other two endpoints. The token is generated using the user’s email and password.GET /search: This endpoint is used to search the chemical compounds data in the database.GET /compound/{id}: This endpoint is used to retrieve detailed information of a specific compound.
Authentication is required for accessing the /search and /compound/{id} endpoints. The user must provide a valid bearer token in the Authorization HTTP header.
Generating the access token
To generate the access token, use the POST /token endpoint. The following parameters should be included in the request body:
email: The email address of the user which has been used to set up an account via the MQS Dashboard.password: The password of the user.
See Tutorial I for a code example.
Searching for compounds
To search for compounds, use the GET /search endpoint. The following parameter should be included in the request URL:
q: The search query string.
One can optimize the search query string by making use of the MQS Dashboard search interface to test the query string by seeing which kind of molecules are found and how to further refine the query string.
For example if you would like to search for the chemical class of polychlorinated biphenyls (PCBs) you can start with typing “polychlorinated biphenyls” or “PCB” in the search field.
Retrieving a compound datasheet
To retrieve the full datasheet of a single compound, use the GET /compound/{id} endpoint. The following parameter should be included in the request URL:
id: The ID of the compound.
The differentiation between the two endpoints GET /search and the GET /compound/{id} can be exemplified with imagining how the search dashboard interface is being used:
The search field element of the dashboard makes use of the GET /search method while when a user clicks on a specific compound, the GET /compound/{id} method is applied.
Dashboard search field query
The search query field of the dashboard allows to tailor your search queries first before retrieving all the data of the molecules. As with many other search engines one can make use of operators (AND, OR, NOT) and wildcards (*, #, ?, %) to refine your search query to retrieve a search result with only a set of structures of interest.
For example to search for polychlorinated biphenyls (PCBs) one can search for the term:
“PCB”
but will then also retrieve a search result with structures including S, N, or O atoms.
A refinement of the search string to:
“PCB AND C12* NOT formula:*S NOT formula:*N* NOT formula:*O”
allows to only retrieve two rings connected to each other and with Cl atoms bounded with the C-ring atoms in different configurations.
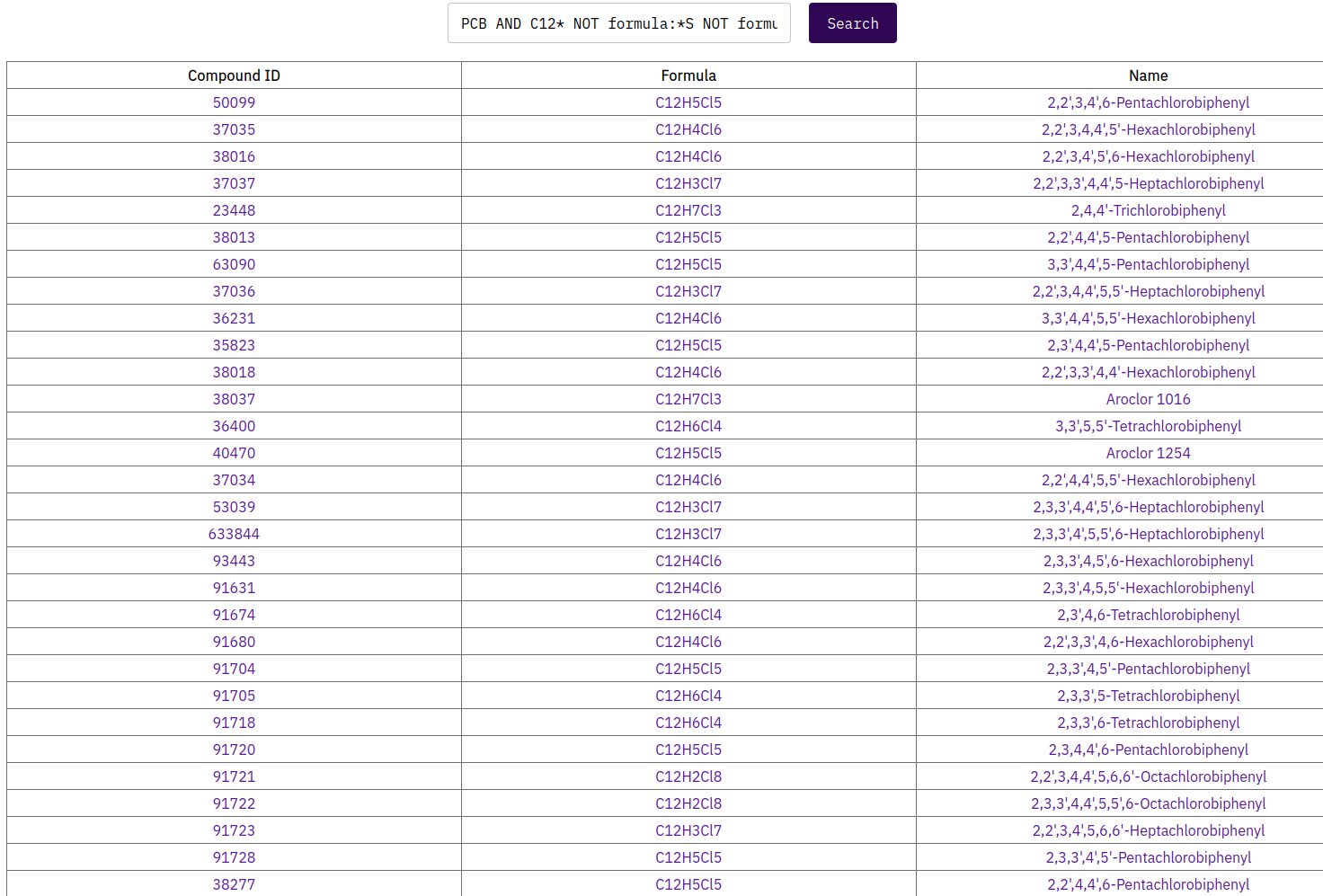
Figure 3: Search query results in MQS Dashboard
Tip: If you are not satisfied with a search because a SMILES string gives you for example only a single atom as the top result or the exact match does not show up at the top, then try to make use of double quotation marks enclosing your SMILES string you are trying to search:
“CC(=O)OC1=CC=CC=C1C(=O)O”
instead of searching for the SMILES without quotation marks:
CC(=O)OC1=CC=CC=C1C(=O)O
try it yourself and see how the results differ.
MQS Database: Currently available data sets (as of July 2023)
The current beta release of the MQS database holds the PubChemQC PM6 and QMugs data sets where the PubchemQC PM6 calculation pipeline has been based on the Pubchem database to screen all available molecular structures whereas the QMugs pipeline makes use of the ChEMBL database.
We refer to both publications of the individual data sets to understand how the data has been generated and what kind of valuable data they hold. Here a comparison table between PubchemQC PM6 and QMugs:
| PubchemQC PM6 | QMugs | |
|---|---|---|
| Data mining source | Pubchem database | ChEMBL database |
| Semi-empirical method | PM6 | GFN2-xTB |
| Software applied | NWChem | RDKit, xtb, PSI4 |
| Availability of conformers data | No | Yes |
| Availability of different electronic states | Yes | No |
| Number of molecular structures | 221.190.415 | 685.917 |
The Search UI & API allow to search through both data sets at the same time via the following identifiers:
|
|
One can also search through each individual data set and zoom further into the schema of the data sets to evaluate what can be searched for.
The PubchemQC PM6 data set holds the following data:
|
|
And the QMugs data set consists of:
|
|
See also these tables from the QMugs paper to get an overview of the data:
https://www.nature.com/articles/s41597-022-01390-7/tables/3
https://www.nature.com/articles/s41597-022-01390-7/tables/4
The user interface of the MQS Dashboard provides an overview of the QMugs data if available for the specific molecule:
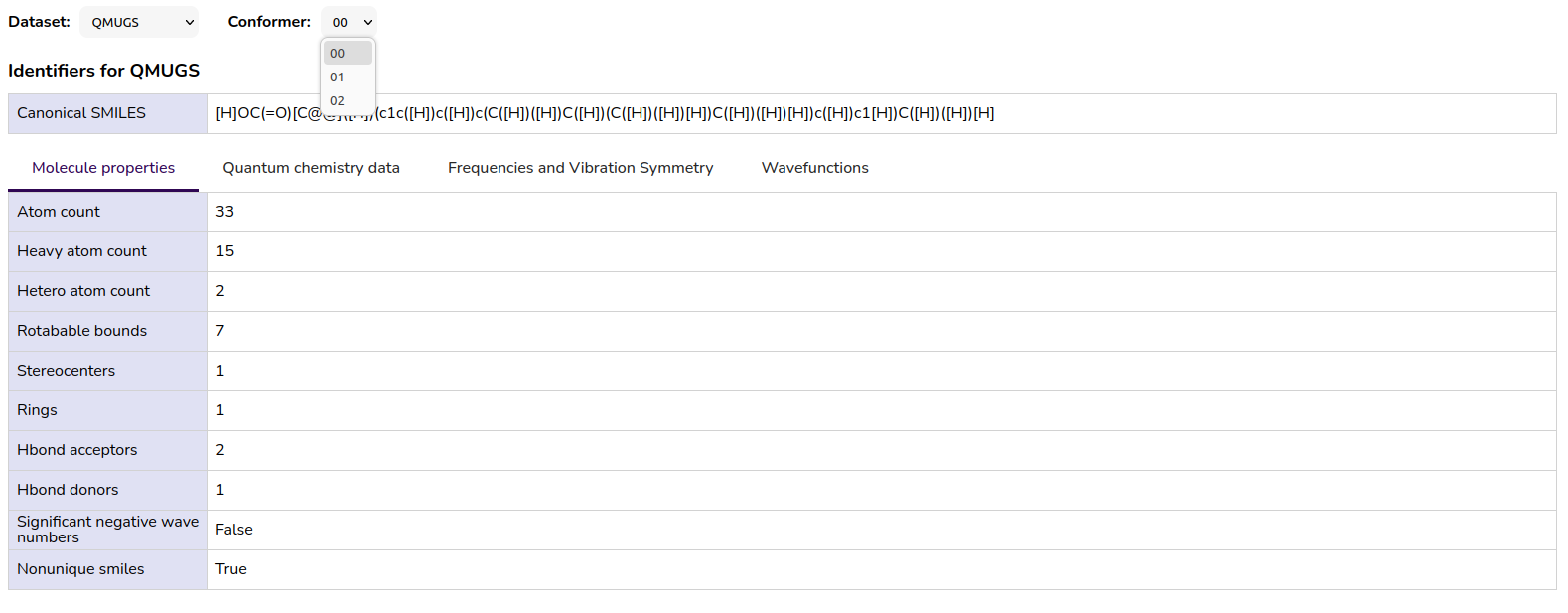
Figure 4: Tabs and table view for QMugs data in the MQS Dashboard
Sometimes you will not find the possibility to select the QMugs data in the dropdown because only PubchemQC PM6 data is available for this specific molecule. Or vice versa.
The available data view for the PubchemQC PM6 dataset can be seen in the following figure:
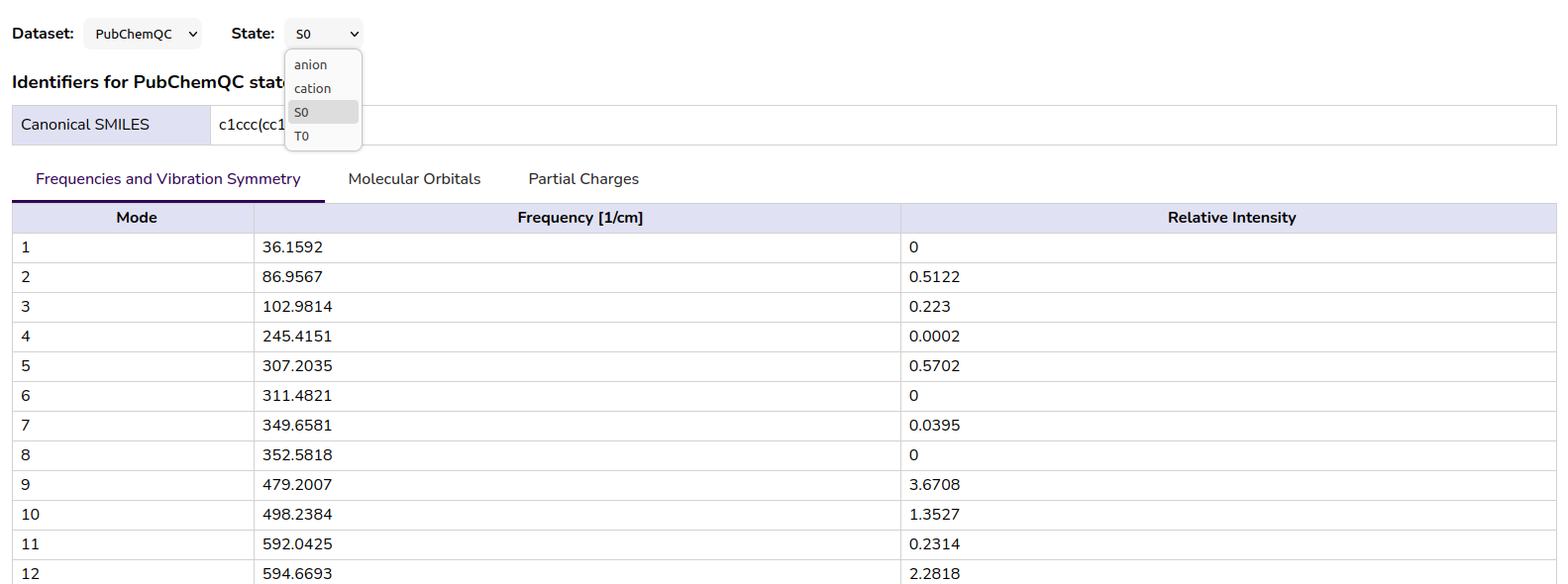
Figure 5: Tabs and table view for PubchemQC PM6 data in the MQS Dashboard
HOMO-LUMO gap data from the PubchemQC PM6 and QMugs data set
Similar to PubchemQC PM6, QMugs holds HOMO-LUMO gap data calculated via the gfn2 method and with DFT. In the following code snippet you can see how to retrieve HOMO-LUMO gap data from both data sets. QMugs contains property data for different conformers (local minima on the potential energy surface of the molecule) whereas PubchemQC PM6 holds data for different states.
|
|
PubchemQC PM6 holds HOMO-LUMO gap data for different molecule states (anion, cation, singlet state, triplet state) which one can select after having selected the “pubchemqc_pm6” data set. It is important to be aware of that not all molecules hold data for every state. In the original PubchemQC PM6 schema this is reflected by the available entries which can be selected via [0], [1], [2], [3]. But these are not fixed to the individual states. Some molecules might only have entries for [0] and [1], where [0] would hold the singlet state data but [1] could hold any other data (anion or cation or triplet state). In the MQS database we have fixed the states data to non-numeric entries ([‘singlet state’], [‘anion’], [‘cation’], [‘triplet state’]).
The above example is also convenient for comparing data points from different data sets. This will be exemplified in our next blog article.
The ChEMBL and SureChEMBL identifiers combined with the retrieval of wavefunction files in the QMugs data set
The ChEMBL identifier has been added to the search IDs which can be used for screening compounds in the MQS database via the search query field in the dashboard or the API. The current supported ID set for searching through the database is summarized in the following:
|
|
For more information about the ChemBL identifier, we recommend the following blog article: http://chembl.blogspot.com/2011/08/chembl-identifiers.html
One has to be careful when searching via for example “chembl*” because one will also receive results where molecules in their synonyms list have a SureCHEMBL (SCHEMBL) identifier. Therefore it is best to search directly with chembl_id in the search string: “chembl_id: chembl*”
Each molecule in the QMugs dataset has up to three conformer structures and is linked to one ChEMBL identifier in the database. To understand the terms “conformation” and “conformer” we quote from the QMugs paper:
“In chemical terminology, the term “conformation” refers to any arrangement of atoms in space, whereas “conformer” refers to a conformation that is a local minimum on the potential energy surface of the molecule.” [40]
Here an example to retrieve the total energy [E_h] data from the QMugs data set with the ChEMBL and SChEMBL identifiers:
|
|
E_h is the total energy in units of Hartree and can be converted to other energy units. Here a convenient converter provided by the Weizmann Institute in Rehovot (Israel): https://www.weizmann.ac.il/oc/martin/tools/hartree.html
And here a conversion overview provided by the National Chiao Tung University (NCTU) in Taiwan which was merged with the National Yang-Ming University to the National Yang Ming Chiao Tung University (NYCU): http://wild.life.nctu.edu.tw/class/common/energy-unit-conv-table-detail.html
The QMugs data set also holds wavefunction related information and are stored as .tar.gz files. In the Dashboard one can download the wavefunction files by clicking on the respective button within the selected tab view:

Figure 6: Download button provided in the MQS Dashboard UI for wavefunctions from the QMugs dataset.
The following three steps can be followed to download the wave function files via the MQS API:
-
Search for a compound and retrieve the compound ID
-
Look up the download URL for the specific compound ID and
-
Download the wavefunction .tar.gz file with the URL, unpack it and look at the content of the file directly in your terminal
The following Python code snippets implement these three steps.
Step 1:
|
|
Step 2:
|
|
Step 3:
|
|
The properties which can be retrieved from the wave function files are listed in Table 3 of the QMugs paper:
https://www.nature.com/articles/s41597-022-01390-7/tables/4
All Jupyter notebooks and Python scripts are available in the JupyterLab environment with the “Quantum C^n” subscription (formerly named “Basic”) of the MQS Dashboard (https://dashboard.mqs.dk). C stands for chemistry and computing. n stands for the exponential advantage one could possibly retrieve with quantum computing for quantum chemistry applications.