Getting started with the MQS Search API - Part 1: Introduction
MQS Documentation
This blog is now defunct. Head over to https://docs.mqs.dk
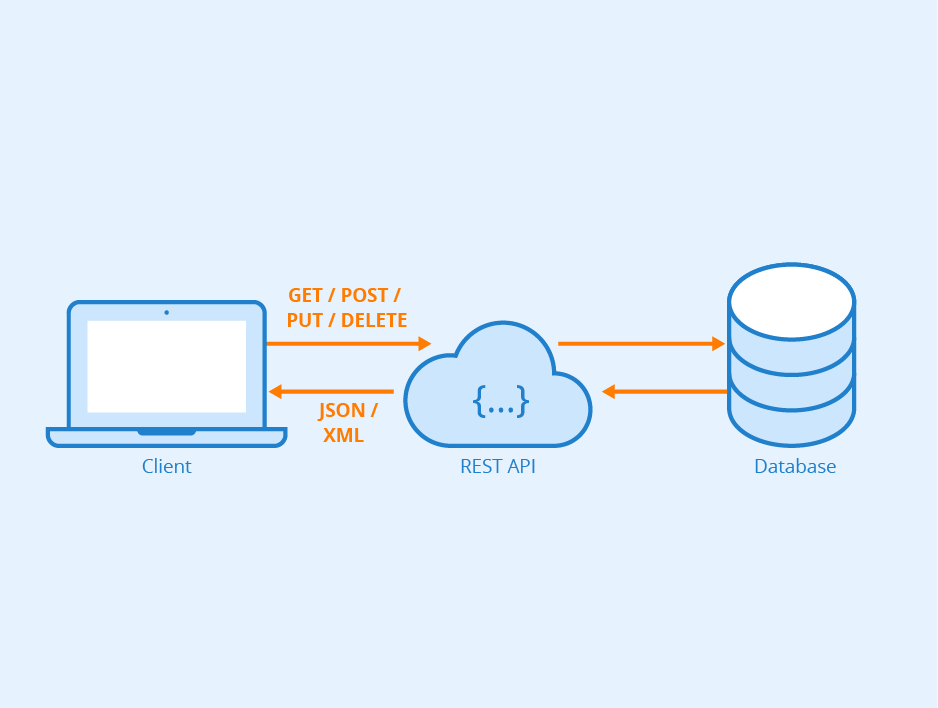
Figure: REST API - Author: Seobility - License: CC BY-SA 4.0
Microservices and containers
Right from the start when MQS was founded we defined a highly modular and microservice based architecture for our cloud software stack. The reasoning behind this decision was that due to the nature of a start-up company we wanted to remain flexible with our portfolio of algorithms and tools. For that reason we containerize the components of our stack (algorithms, database, API, UI) as much as possible and combine them for the needed pipelines/workflows. The first microservice we are releasing as a beta version is our Search API which connects to our database system. The database holds data of molecules and will be extended over time with additional data sets published under free licenses such as the GPL, MIT or Creative Commons licenses.
We have made our current database freely accessible through the user interface within the MQS Dashboard and on a subscription basis via the MQS Search API.
In the following we are describing how you can make use of the Search API when you have subscribed to the basic plan. You can find the subscription overview at the top right of the user menu in the dashboard.
Authentication to the MQS Search API
To authenticate via the Search API you need to make use of your email address which you have used for logging into the dashboard.
In the below code you can see where you have to provide your email address and your password in the credentials dictionaries’ key-value pairs “username”:“YOUR_EMAIL” and “password”: “YOUR_PW”.
|
|
By running the above Python script a token is retrieved from the API post request. This token is being stored in the headers dictionary which will be utilized for any other request to the API.
The last lines in the above script showcase how you can search for a molecule in the database with an isomeric SMILES string.
When printing the retrieved information with the print() function one can see that the API has sent back data and it is being resolved as a dictionary by the requests package, due to the applied json() function. The data dictionary holds a lot of nested information. You can run the above example in this Python script or Jupyter Notebook which we have compiled covering the examples in this tutorial.
As you will see, the last print() function gives an overview of the complete data set for the ethane (CC) molecule.
Get individual properties of molecules
The next code example shows how one can retrieve specific information of a molecule such as the main title (main compound name) of the molecule, the number of atoms, the geometry optimized xyz coordinates of the individual atoms of the molecule, the vibrational spectra and orbitals information.
Important to acknowledge is that first the molecule id has to be retrieved from the data one has obtained from a previous request. The below code starts where the last code example has ended:
|
|
As you might have now noted, we have introduced a new request endpoint: {api_url}/compound and made use of the id retrieved from the data which we in turn requested from the {api_url}/search endpoint.
We are interacting with the API as we would do in the user interface: first we search for a compound and then we request further information on a searched molecule.
How to obtain detailed information could be quite confusing in the beginning since one has not yet seen the complete schema of the database. For a start we will go through the five examples depicted in the above code: title, number of atoms, 3d coordinates, vibrational spectra and orbitals.
To access the title we can make a first level look-up: data["title"]. That was easy.
The number of atoms (atom count) is currently stored in a specific data set which the database holds and therefore one needs to first reference the data set and then the state of the molecule (e.g. 0: anion, 1: cation, 2: S0, singlet state 3: T0, triplet state), followed by ['atoms']['elements']['atom_count'].
It is important to acknowledge that the PubchemQC PM6 data set holds information for various states and sometimes no data for a specific state is available. Therefore one can not just automate going through the index assuming that the indices 0, 1, 2 and 3 hold information about the anion, cation, singlet and triplet state. If for a molecule no information is available for the cation state, then index 1 would hold information about the singlet state and index 2 about the triplet state. In the last section of this tutorial we show how one can easily check and automate how to high-throughput screen through specific state information.
Back to the above code example: data["pubchemqc_pm6"][0]["atoms"]["coords"]["3d"] let’s one access geometry optimized 3d coordinates data which is being stored in a numpy array and reshaped so that one can view the xyz columns element by element (rows) in the output.
Now it should be clear that one needs to have some idea how the data set has been stored in the database and for this the schema is of importance for a user. In the following you can see the full schema of the PubchemQC PM6 data set which helps to request specific data from the API:
|
|
How the vibrational spectra and orbitals information have been referenced in the above code example can be comprehended by comparing the Python code with the schema.
Other identifiers to search with
In the previous example a search was performed with a SMILES string. But there are several other identifiers you can make use of for searching the database. For example, the Pubchem CID, the main name title (main compound name) of the molecule, synonyms of the molecule, InChi and InChi key.
Here a complete overview what one can use as search queries via the API:
|
|
Currently the database consists of the PubchemQC PM6 data set(pre-print arXiv paper).
pubchemqc_pm6.state and pubchemqc_pm6.openbabel_canonical_smiles show how you can go directly into a data set to search for example via the available molecule states (e.g. S0, anion, cation) or for the openbabel_canonical_smiles which have been generated in this data set.
In the future more data sets will be available and the API will allow you to seamlessly search with the main identifiers through all data sets.
Get sets of molecules information
The following Python script example shows how one can high-throughput screen via the API to retrieve sets of molecule information. We apply a list comprehension instead of a for loop to iterate over API requests. The authentication procedure is now defined with a function and the main part shows how the ids of the found compounds are requested from the Search API and stored in the id set list. The queries list is generated by iterating over carbon chains from C2 to C10 and different identifiers are utilized. Isomeric SMILES, CID, InChI key and CAS work the best to receive an exact match with the respective identifier.
|
|
State look-up
The PubchemQC PM6 data set indices for the states can change because some compounds do not hold data for all states.
The following code snippet shows how one can read out the data from different states for a set of molecules:
|
|
Before designing a high-throughput pipeline via the MQS Search API and tailored scripts, one can also make use of the MQS Dashboard Search UI to easily check what kind of data one can find in the database.
For example you can use the dashboard search field to screen for all available S0 state data by typing pubchemqc_pm6.state:S0 into the search input field.
Jupyter notebook and Python script
You can find a Jupyter notebook and a Python script implementation of the above examples in our dedicated github tutorials repo.