The Era of the Digital Pharma Lab
Behind all new drugs being released to the market is a long research journey. [1, 2] A drug discovery process starts with identifying the target in the human organism on which a new molecule should have a therapeutic effect on. After finding promising lead candidates, scientists hunt for more information about the new active pharmaceutical compounds through trial and error experiments (Figure 1). Finally, a drug product needs to be designed in the form of a tablet, injection or ointment (skin cream). This journey is an expensive, time- and material-consuming endeavour, and a heavily regulated process. Therefore pharma R&D units are in need of tools which can speed up their efforts to identify new drug molecules and to optimize the product formulation steps.
 Figure 1: Drug discovery and development pipeline at the pre-clinical and clinical stages.
Figure 1: Drug discovery and development pipeline at the pre-clinical and clinical stages.
The Eroom effect
In 1965 Gordon Moore observed and extrapolated a trend how the capabilities of the semi-conductor industry would develop for the upcoming decades with respect to the number transistors implemented on a microchip. This trend follows exponential behaviour with the number of transistors doubling every two years. [3] In 2012 Jack Scannell, Alex Blanckley, Helen Boldon and Brian Warrington discussed the reversed efficiency of R&D efforts of the pharma industry. [4] They termed this trend profile as the Eroom (Moore in reverse) effect. In this article we will focus on the Eroom trend profile and if in-silico (computer) simulations can contribute to break the inefficiency of pharma R&D pipelines. A Boston Consulting Group (BCG) report, with Jack Scannel being a co-author again, has shown with a statistical analysis that a trend reversal of the Eroom curve can be seen from 2010 on. [5] This is caused by three advancements highlighted by the following three points:
- Genetic data sourcing which helps to develop therapeutics for small patient populations which was not possible during clinical development in the past.
Here in vitro discovery (experiments with isolated organisms) has become an important part of the pre-clinical and clinical drug development pipeline where DNA library technologies and phage display are important parts of the tool set when it comes to antibody based drug discovery for example. [6]
- Decision-making processes during pre-clinical and clinical drug development are being improved to rule out human bias or other deficiencies.
To highlight here is that machine learning and Bayesian statistical analysis can improve and automate to a large degree the analysis of the produced information during the drug development process.
- A paradigm and/or workflow shift at regulatory authorities.
It is not completely clear what these shifts are caused by, except that a shift to small patient populations with higher success rates for rare diseases can be one reason to more likely obtain approval than before. We assume that also better statistical analysis methods have been implemented and are used by regulators. The FDA for example maintains a GitHub repository for different tools and platforms which can help with “the communication and collaboration with data and computational results using biocompute paradigm”. [7,8]
We believe that the above points will be further improved and extended with quantum chemistry simulations and artificial intelligence (AI) / machine learning (ML). AI/ML will make use of large structured data sets, quantitative biology and next-generation sequencing techniques to further reverse the Eroom effect. Quantum chemistry models will provide more insight into binding behaviours of drug molecules to specific targets and property predictions of protein based therapeutics such as antibodies. Several modelling applications which can be connected to quantum chemistry simulation results are for example transdermal drug delivery modelling, drug formulation optimization or drug manufacturing models.
Combined with the availability of high performance computing infrastructure in the cloud, researchers and engineers are not dependent anymore on academic institutions with large facilities and capabilities to run such quantum chemistry simulations. We observe actually a shift to industry driven research with deep pockets to run very expensive AI models which academic institution can not cover from a budget perspective without investing in a dedicated GPU infrastructure. One example is the training recipe of the AlphaFold algorithm which is well documented in Table 5 in the supplementary material of the respective paper. [9,10] The model took more than 20 days to train and the cost can be back-calculated from the tensor processing unit (TPU) pricing. The costs were at least over 1 mio US$ solely for training the neural network. [11] To counter-act this trend in academia, different initiatives are being pursued in order for universities to get access to the needed computational power in the future. [12]
The automated deployment of quantum chemistry packages to cloud provider resources and the ease of use of such models will allow researchers to gain better information before going into the laboratory to conduct experiments. This kind of scientific work has to be combined with a consistent software development life cycle (DevOps and MLOps) to achieve efficient and maintainable implementations.
Quantum chemistry
Quantum chemistry theory is built on the fundamental laws of quantum physics. [13,14,15] An important advantage is that these methods rely only on a minimal set of measured data, which means that researchers spend less time performing long and costly laboratory experiments. Therefore, incorporating computational modelling achieves a significant acceleration in the drug development process. Although computational quantum chemistry has been used as a supportive method over the last couples of decades, it is increasingly becoming an inherent part for pharma R&D research.
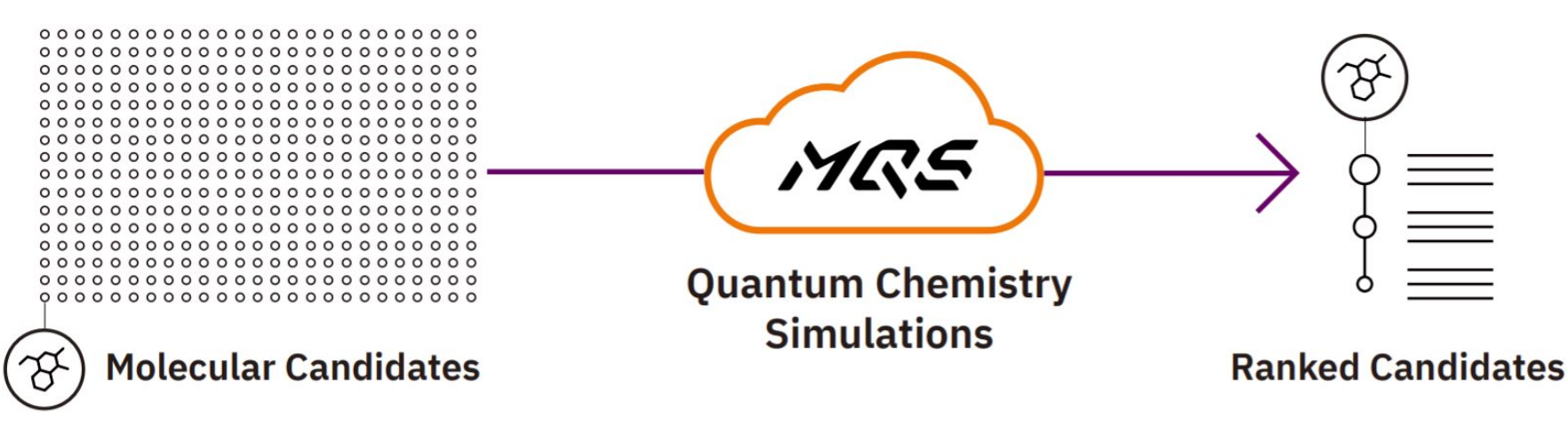 Figure 2: High performance quantum chemistry simulations in the cloud for high throughput analysis of chemical structures.
Figure 2: High performance quantum chemistry simulations in the cloud for high throughput analysis of chemical structures.
The reason for this development is the availability of more powerful computers. Either in form of local machines (desktop and laptop computers) to prototype and test algorithms, or to deploy as an algorithm in production in the cloud (connected distributed server networks). Several users can then make use of the developed algorithms for more demanding calculations with these distributed server networks. As a result, pharma companies are currently undergoing a digitalisation transformation process in order to integrate modern computational pipelines and computational modelling methods. [16]
Let’s break down the previously mentioned drug development pipeline in detail.
By applying quantum chemistry, initial support can be given to find new active pharmaceutical ingredients (APIs) and their interactions with target proteins in the human body. Once promising molecules have been identified, one needs to characterize all needed physico-chemical properties of these molecules such as solubilities in solvents or distribution (partition) coefficients among various fluids in the human body. This step is of importance for the design of the optimal final product form. One needs to know the solubility of the API in solvents such as water, ethanol, polymeric matrices as well as gastric or intestinal fluids to understand how the drug will distribute in the human body. These kind of solubility and viscosity predictions can be obtained with continuum solvation models and group contribution models where quantum chemistry calculations can be applied.
Implicit (continuum) solvation models predict the solubility based on the electrostatic nature of the chemical system. The solvent is modelled as a continuous medium around the solute (compound which is being solved in the solvent), rather than trying to model each single solvent molecule around the solute as performed with explicit solvation models such as molecular dynamics (MD). Quantum chemistry models are applied to calculate the charge distributions of the solute cavity in the specific solvent. For the same solute, but different solvents, this electric density charge profile will change due to the varying polarity of the solvent. Those pre-paramterized solvation models allow to predict solubilities without the need of conducting thermodynamic experiments for new molecules. The parity plot below shows how well our MQS solvation model performs when applying it to bio-active compounds in single solvent and mixed solvent systems.
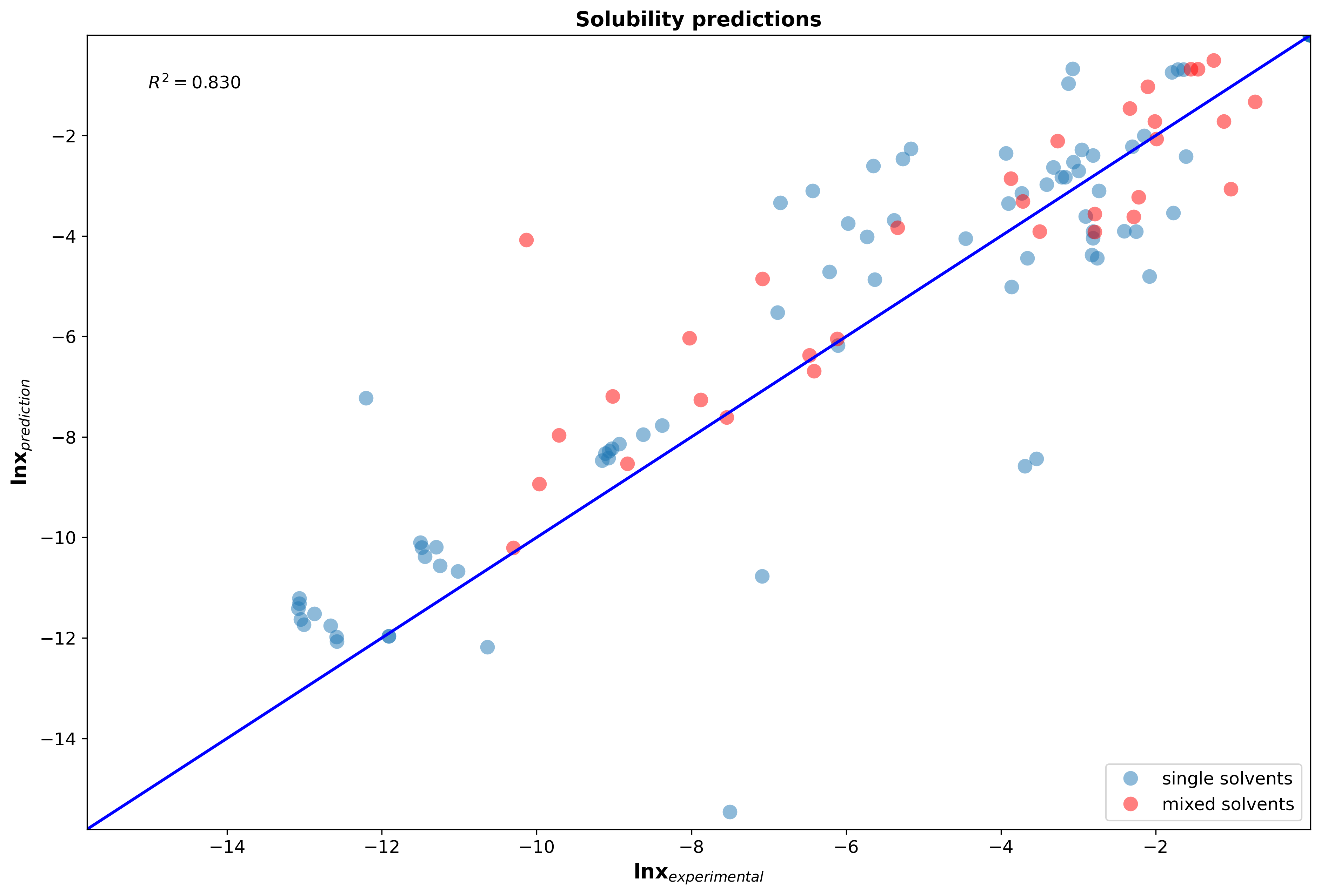 Figure 3: Parity plot of solubility predictions of 132 systems (including hydrocarbons, small drug like molecules, peptides, esters, ketones, alcohols, polymers, glycerides) in single solvent and solvent mixtures at different temperatures and compositions.
Figure 3: Parity plot of solubility predictions of 132 systems (including hydrocarbons, small drug like molecules, peptides, esters, ketones, alcohols, polymers, glycerides) in single solvent and solvent mixtures at different temperatures and compositions.
The other mentioned class of predictive property models, namely group contribution (GC) models, dissect molecules into pre-determined smaller groups. These individual groups are assigned a GC value for the specific property of interest by applying a regression method. The drawback with GC methods are that an experimental data set is needed to create the model. If this data set lacks compounds which embed the specific structural group, then the model will not be able to predict a value for a compound of interest which needs a GC value assigned to this specific group. Here, quantum chemistry based prediction models can be of use to generate a data set to re-fit the GC model to either fix the issue of missing GC values or to tune the parameter set of the regression model for a specific chemical class, e.g. polymers.
Quantitative structure activity/property relationship (QSAR/QSPR) models are similar to group contribution methods where an experimental data set is fitted with the help of molecular descriptors. The group count and type of group (assigned with a GC value) were the descriptors in case of a GC model. QSAR/QSPR models can be fitted with a variety of descriptors, also quantum chemical ones, to describe a set of molecules. Table 1 shows how different property models can be retrieved with different methods and for which applications these predictive models are important.
Table 1: Properties of chemicals and suitable prediction models for applications in drug development and manufacturing.
| Property | Method/Model | Example applications |
|---|---|---|
| Partition coefficient | Implicit solvation | Separation processes in manufacturing |
| Solubility | Implicit solvation | Formulation optimization in product development |
| Viscosity | GC | Final formulation optimization in product development |
| Toxicity | QSAR/QSPR | Filtering of molecules during drug discovery and development phase |
| Absorption, Distribution, Metabolism, Excretion (ADME) | QSAR/QSPR | Filtering of molecules during drug discovery and development phase |
The prediction accuracy of property models can be further improved by leveraging the accuracy of quantum chemistry methods. These methods and algorithms become very computationally demanding if performed for large molecules, e.g. increasing size of peptides. There are several propositions how to speed up and refine quantum chemistry calculations. On the hardware side this can be realized with the use of massive parallel threads on CPUs or the utilization of GPUs and FPGAs when machine learning models are involved such as neural networks. These capabilities can accelerate computations to a significant degree and MQS provides highly tailored algorithms on CPUs, GPUs and FPGA designs in the MQS portfolio.
Quantum computing
The hype traction but also the promising developments of quantum computing are in full swing and at MQS we are preparing to have this additional computing unit integrated within our technology stack. [17,18,19,20,21,22]
Important to understand is that quantum computing will be feasible for some use-case applications in the near term future, luckily quantum chemistry is one of them, whereas other applications are still at least a decade away from adopting quantum computers as a computational device.
Quantum computing is a new way of computing that relies on using the laws of quantum physics as a tool for performing calculations in an extended state space of 2 to the power of n, where n is the number of available qubits on the quantum compute device. The chemical interactions that occur in nature are governed by these laws, which is why quantum computing is especially suitable for performing quantum chemistry simulations. The components in today’s conventional computers, phones, and tablets are not designed for this type of computation. In fact, not even the most advanced supercomputing infrastructure in the world has this sort of capability, which is why new technology is being developed, to produce quantum computers specifically tailored for this task. The new hardware has an entirely different architecture, which is why existing simulation software cannot be directly ported to the new devices. This is where MQS contributes with developing specialized algorithms and a software platform that connects to these devices by making the capabilities of quantum computing accessible for those in need of quantum chemistry simulations.
One of the algorithm classes which will be valuable for quantum chemistry calculations are variational quantum eigensolvers (VQEs) which exist in various modifications. In comparison to other algorithm classes which are useful for quantum chemistry calculations, e.g. quantum phase estimation (QPE), VQE algorithms can be solved on noisy intermediate-scale quantum (NISQ) devices which are already being launched on the market by companies such as IBM, Rigetti, IonQ and Quantinuum. These quantum computers come with a low number of qubits, short coherence times and high error rates. At MQS we are developing a consistent and rigorous benchmarking framework for VQE algorithms while solving various use-case applications. Other exciting news will be announced about our quantum computing efforts in the upcoming months including a research grant project with several partners involving photonic quantum computing.
Our efforts analyze the whole spectrum of available computing units (CPUs, GPUs, FPGAs and QPUs) in a holistic approach to dissect methods and pipelines into individual parts and divide them up between these processing units. We believe that the future of quantum computing will be of a hybrid nature, on the hardware side, as well as on the software side. We see more and more research being published which combine different quantum algorithms (VQE + QAOA) together with classical algorithms. [23] To modularize the development efforts of a software company it will be critical to have a well thought out and implemented testing system in place to benchmark the individual algorithms on different devices. And to communicate the efficiency in comparison to other implementations to the researchers and engineers working with these algorithms. Figure 4 shows with a simplified mental model how to visualize the advantage of quantum algorithms over classical implementations. We recommend to read [24] for a more in-depth discussion about the classification of the computational advantages between classical and quantum algorithms.
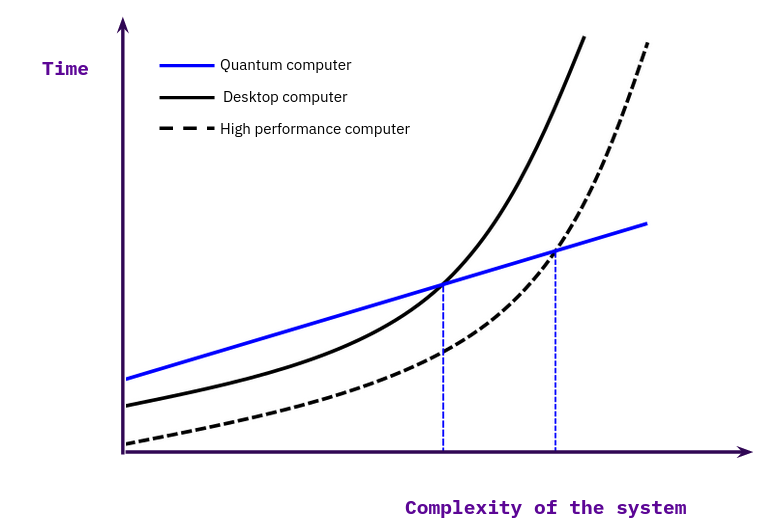 Figure 4: Compute time over complexity size for classical and quantum computations. Adapted from Fig. 3 in [24].
Figure 4: Compute time over complexity size for classical and quantum computations. Adapted from Fig. 3 in [24].
With many governments and corporations investing heavily into research and development, the quantum computing market is projected to have a 56% compound annual growth rate during the next decade. [25] The organizations that will capture the most value from this market are those who use available computing power in the most efficient way. Companies involved in drug discovery, formulation design, materials design, chemical process design, biochemical engineering, and other areas will all benefit from the accelerated chemistry simulations, made possible with the advent of quantum computing. Therefore it is important that companies start to train their researchers and engineers in need of those tools.
Molecular Quantum Solutions
To conclude and summarize what we develop at Molecular Quantum Solutions (MQS), we tried to present the scope of the technology we are working with to predict relevant properties of for example APIs in solvents based on quantum chemistry methods. The only input for such calculations performed on a powerful computer is the 3D structure of each molecule of interest and a few fundamental physical properties, which are all easily obtainable. The output from the calculation is then forwarded to the mathematical property models to predict the macroscopic properties, such as solubility. It has been demonstrated that such models provide qualitative results for high throughput screening and the tool provided by MQS furthermore provides the quantified prediction uncertainty. Hence, properties for thousands of molecules can be predicted faster than measurements can be done by any laboratory. Figure 5 shows a bottom up calculation of a research study where a set of 20 solvents were evaluated for a specific API from a pharma company. The fully automated in-silico MQS pipeline allowed to receive needed results to formulate the API in a solvent mixture and progress further in the pre-clinical phase. The advantage scales with the number of such studies and the number of compounds being analyzed with the MQS platform.
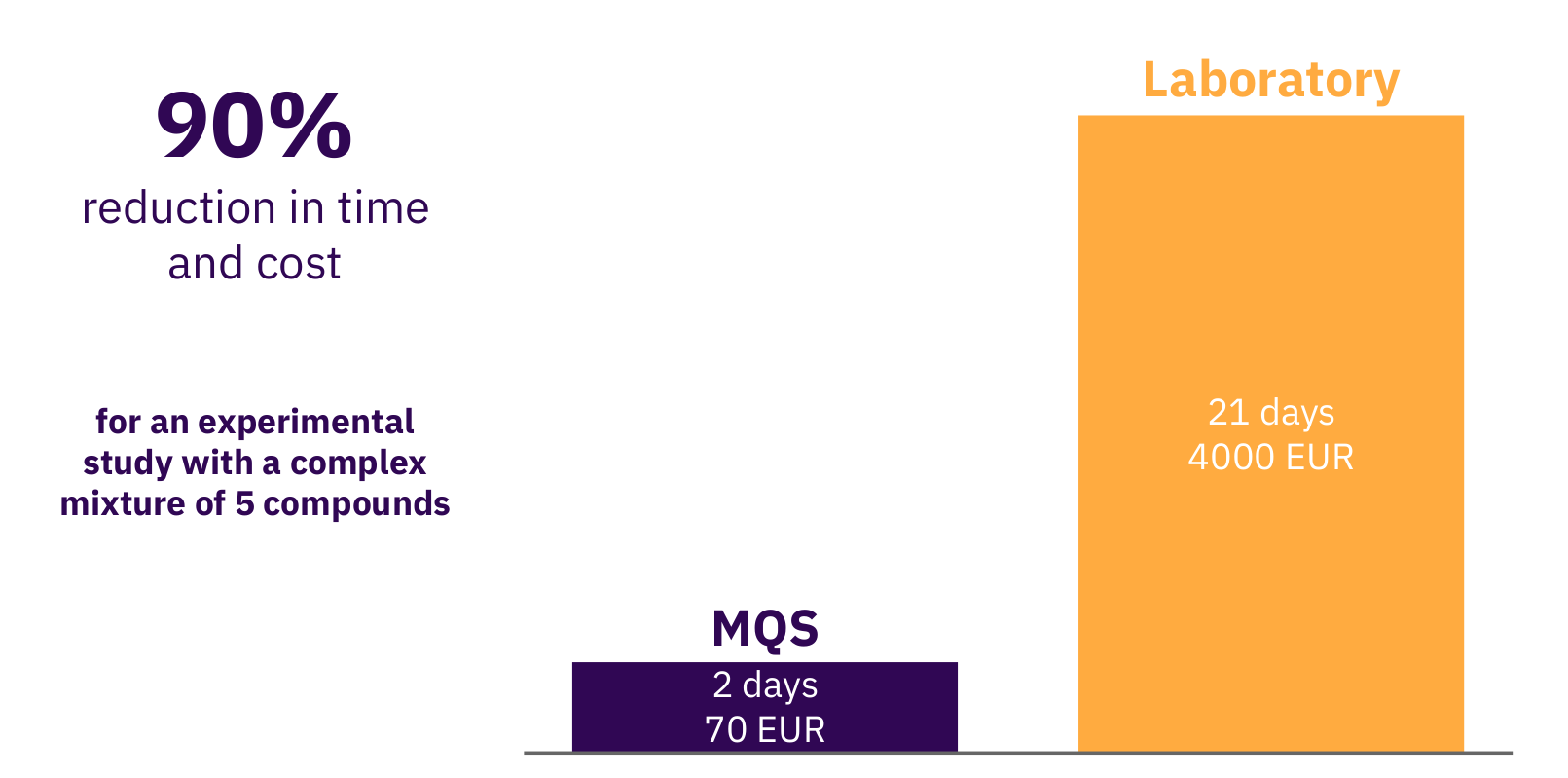 Figure 5: Research study with a pharma company conducted by MQS for drug formulation analysis.
Figure 5: Research study with a pharma company conducted by MQS for drug formulation analysis.
Our strategy is to focus our market entry efforts with pharma companies to set the highest boundaries for our software. We design it compliant to the relevant ISO frameworks (ISO9001, ISO27001), the FDA framework 21 CFR Part 11 and conforming GxP (GAMP 5) guidelines. Figure 6 visualizes the capabilities of the MQS dashboard and application programming interface which can be integrated into the existing tool stack of a company’s infrastructure. But the prediction capabilities of our models are not limited to pharma related applications. Biotech and chemical companies can profit likewise from these simulations when use-case applications within their business portfolio and R&D efforts have been identified.
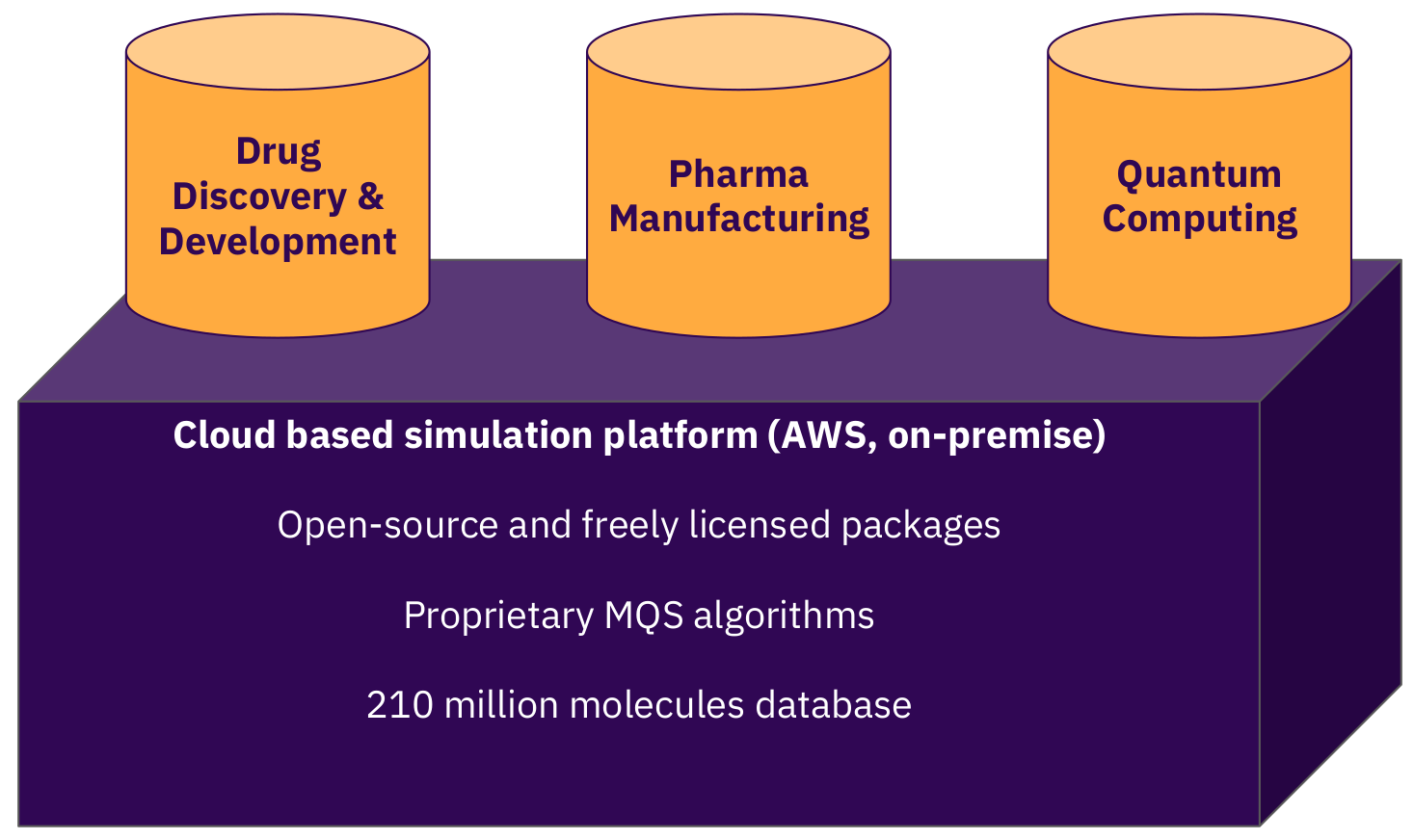 Figure 6: The MQS cloud platform for advanced calculations for the pharma, biotech and the chemicals industries.
Figure 6: The MQS cloud platform for advanced calculations for the pharma, biotech and the chemicals industries.
Please reach out by using our contact form on our webpage and let us know if you have any questions or topics/use-cases your are highly interested in.
References
[1] https://www.statista.com/statistics/309471/randd-spending-share-of-top-pharmaceutical-companies/
[2] Sandra Kraljevic, Peter J Stambrook and Kresimir Pavelic; “Accelerating drug discovery”; EMBO Rep 2004 Sep; 5(9):837-42; https://doi.org/10.1038/sj.embor.7400236
[3] Gordon E. Moore; “Cramming more components onto integrated circuits”; Electronics, Volume 38, Number 8, April 19, 1965; https://newsroom.intel.com/wp-content/uploads/sites/11/2018/05/moores-law-electronics.pdf
[4] Jack W. Scannell, Alex Blanckley, Helen Boldon and Brian Warrington; “Diagnosing the decline in pharmaceutical R&D efficiency”; Nat Rev Drug Discov 11, 191–200 (2012); https://doi.org/10.1038/nrd3681
[5] Michael S. Ringel, Jack W. Scannell, Mathias Baedeker and Ulrik Schulze; “Breaking Eroom’s Law”; Nature Reviews Drug Discovery 19, 833-834 (2020); https://doi.org/10.1038/d41573-020-00059-3
[6] Pietro Sormanni, Francesco A. Aprile and Michele Vendruscolo; “Third generation antibody discovery methods: in silico rational design”; https://doi.org/10.1039/C8CS00523K
[7] FDA launches HIVE Open Source: a platform to support end to end needs for NGS analytics; https://github.com/FDA/fda-hive, https://doi.org/10.1093/database/baw022
[8] BioCompute; https://www.biocomputeobject.org/
[9] John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, Alex Bridgland, Clemens Meyer, Simon A. A. Kohl, Andrew J. Ballard, Andrew Cowie, Bernardino Romera-Paredes, Stanislav Nikolov, Rishub Jain, Jonas Adler, Trevor Back, Stig Petersen, David Reiman, Ellen Clancy, Michal Zielinski, Martin Steinegger, Michalina Pacholska, Tamas Berghammer, Sebastian Bodenstein, David Silver, Oriol Vinyals, Andrew W. Senior, Koray Kavukcuoglu, Pushmeet Kohli and Demis Hassabis; “Highly accurate protein structure prediction with AlphaFold”; Nature volume 596, pages 583–589 (2021); https://doi.org/10.1038/s41586-021-03819-2
[10] Supplementary material of [9]: https://static-content.springer.com/esm/art%3A10.1038%2Fs41586-021-03819-2/MediaObjects/41586_2021_3819_MOESM1_ESM.pdf
[11] Martin P Andersson, Mark Nicholas Jones, Kurt V Mikkelsen, Fengqi You and Seyed Soheil Mansouri; “Quantum computing for chemical and biomolecular product design”; Current Opinion in Chemical Engineering, Volume 36, June 2022, 100754; https://doi.org/10.1016/j.coche.2021.100754
[12] https://blogs.nvidia.com/blog/2021/03/23/ai-supercomputer-sweden/
[13] Yu-hong Lam, Yuriy Abramov, Ravi S. Ananthula, Jennifer M. Elward, Lori R. Hilden, Sten O. Nilsson Lill, Per-Ola Norrby, Antonio Ramirez, Edward C. Sherer, Jason Mustakis, and Gerald J. Tanoury; “Applications of Quantum Chemistry in Pharmaceutical Process Development: Current State and Opportunities”; Org. Process Res. Dev. 2020, 24, 8, 1496–1507; https://doi.org/10.1021/acs.oprd.0c00222
[14] Ola Engkvist, Per-Ola Norrby, Nidhal Selmi, Yu-Hong Lam, Zhengwei Peng, Edward C Sherer, Willi Amberg, Thomas Erhard and Lynette A Smyth; “Computational prediction of chemical reactions: current status and outlook”; Drug Discov Today, 2018 Jun; 23(6):1203-1218; https://doi.org/10.1016/j.drudis.2018.02.014
[15] Scott Aaronson; “Why quantum chemistry is hard”; Nature Physics volume 5, pages 707–708 (2009); https://doi.org/10.1038/nphys1415
[16] Christos Varsakelis, Sandrine Dessoy, Moritz von Stosch and Alexander Pysik; “Show Me the Money! Process Modeling in Pharma from the Investor’s Point of View”; Processes 2019, 7(9), 596; https://doi.org/10.3390/pr7090596
[18] Matt Langione, Corban Tillemann-Dick, Amit Kumar and Vikas Taneja; “Where Will Quantum Computers Create Value—and When?"; https://www.bcg.com/publications/2019/quantum-computers-create-value-when
[19] Jean-François Bobier, Matt Langione, Edward Tao and Antoine Gourévitch; “What Happens When ‘If’ Turns to ‘When’ in Quantum Computing?"; https://www.bcg.com/publications/2021/building-quantum-advantage
[20] Anika Pflanzer, Wolf Richter and Henning Soller; “A quantum wake-up call for European CEOs”; https://www.mckinsey.com/business-functions/mckinsey-digital/our-insights/a-quantum-wake-up-call-for-european-ceos
[21] Matthias Evers, Anna Heid and Ivan Ostojic; “Pharma’s digital Rx: Quantum computing in drug research and development”; https://www.mckinsey.com/industries/life-sciences/our-insights/pharmas-digital-rx-quantum-computing-in-drug-research-and-development
[22] Anna Heid and Ivan Ostovic; “Recalculating the future of drug development with quantum computing”; https://www.mckinsey.com/industries/life-sciences/our-insights/recalculating-the-future-of-drug-development-with-quantum-computing
[23] Qi Gao, Gavin O. Jones, Michihiko Sugawara, Takao Kobayashi, Hiroki Yamashita, Hideaki Kawaguchi, Shu Tanaka and Naoki Yamamoto; “Quantum-Classical Computational Molecular Design of Deuterated High-Efficiency OLED Emitters”; https://arxiv.org/abs/2110.14836
[24] Benjamin A. Cordier, Nicolas P. D. Sawaya, Gian G. Guerreschi, Shannon K. McWeeney; “Biology and Medicine in the Landscape of Quantum Advantages”; https://arxiv.org/abs/2112.00760
[25] V.E. Elfving, B.W. Broer, M. Webber, J. Gavartin, M.D. Halls, K. P. Lorton, A. Bochevarov; “How will quantum computers provide an industrially relevant computational advantage in quantum chemistry?"; https://arxiv.org/abs/2009.12472